Rối loạn chuyển hoá bẩm sinh là gì?Các rối loạn chuyển hóa hoặc các rối loạn chuyển hóa bẩm sinh gây nên bởi sự tắc nghẽn (một phần hoặc hoàn toàn) một con đường chuyển hóa thiết yếu của cơ thể. Các bệnh rối loạn chuyển hoá bẩm sinh được sàng lọc:
I. Rối loạn chuyển hoá bẩm sinh là gì?
Các rối loạn chuyển hóa hoặc các rối loạn chuyển hóa bẩm sinh gây nên bởi sự tắc nghẽn (một phần hoặc hoàn toàn) một con đường chuyển hóa thiết yếu của cơ thể.
Các bệnh rối loạn chuyển hoá bẩm sinh được sàng lọc:
+ Rối loạn chuyển hoá acid amin, chu trình ure
+ Rối loạn chuyển hoá acid hữu cơ
+ Rối loạn chuyển hoá acid béo
+ Rối loạn chuyển hoá carbonhydrate: Galactosemia
+ Rối loạn dự trữ thể tiêu bào: Pompe, MPS, Gaucher, Fabry

(Ảnh minh họa. Nguồn: Internet)
II. Biểu hiện triệu chứng của rối loạn chuyển hoá bẩm sinh như thế nào?
- Có thể xuất hiện ở mọi độ tuổi từ sau sinh tới trưởng thành
- Người bệnh có thể phát triển bình thường cho tới khi có biểu hiện bệnh
- Biểu hiện triệu chứng đa dạng của nhiều cơ quan như tiêu hoá, gan mật, tim mạch, thần kinh, tâm bệnh…. với các nhóm triệu chứng không đặc hiệu sau:
- Giai đoạn sơ sinh:
Bú kém, bỏ bú hoặc nôn
Giảm trương lực cơ
Bất thường hô hấp, ngừng thở
Bệnh não tiến triển hoặc co giật
Bệnh cảnh lâm sàng thường nhầm với nhiễm trùng nặng.
- Ở trẻ lớn hơn:
Nôn mất nước nặng tái phát không giải thích được.
Các đợt giống như đột quỵ
Suy gan và thận cấp
Bệnh lý cơ tim
Bệnh lý não và co giật không giải thích được.
- Các triệu chứng có thể âm thầm:
Chậm phát triển tinh thần hoặc thoái triển
Bộ mặt thô bất thường hoặc bất thường xương
Rối loạn tâm thần.
- Tiến triển tự nhiên của bệnh: người bệnh sẽ tử vong trong các đợt cấp hoặc có di chứng thần kinh nếu không được chẩn đoán đúng và điều trị kịp thời.
III. Nguyên nhân
- Đột biến các gen sản xuất các enzyme, yếu tố đồng vận, protein vận chuyển, receptor của con đường chuyển hoá trong cơ thể dẫn tới tắc nghẽn chuyển hoá.
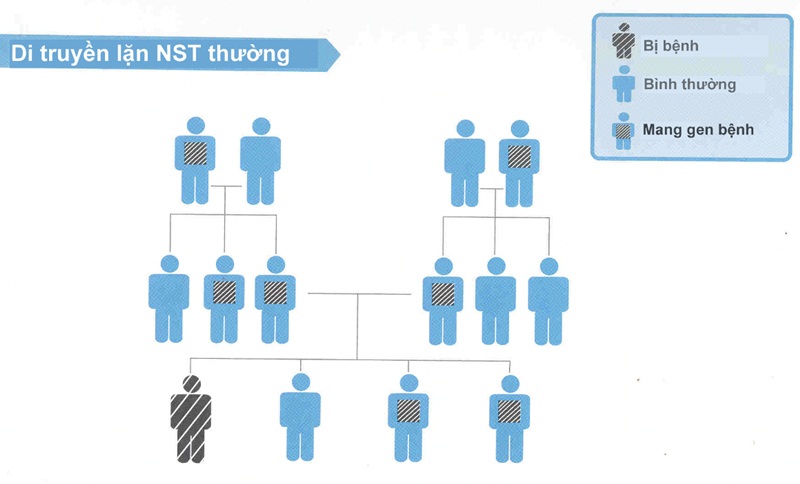
- Đa số các bệnh là di truyền trên nhiễm sắc thể thường. Nam và nữ đều có khả năng mắc bệnh như nhau.
IV. Tỉ lệ mắc bệnh như thế nào?
– Nếu tính riêng từng bệnh, tỉ lệ mắc từ 1: 10,000 – 1: 30,000. Nhưng nếu tính chung 55 bệnh tỉ lệ mắc bệnh từ 1: 2,200 – 1: 7,000.
– Tại Việt Nam: tỉ lệ mắc qua sàng lọc sơ sinh là 1: 3,500.
V. Nhóm bệnh rối loạn chuyển hoá bẩm sinh có điều trị được không?
– Đa số các nhóm là có thể cứu chữa được nhưng phải điều trị suốt đời.
– Nếu được sàng lọc và chẩn đoán bệnh trước khi có triệu chứng xuất hiện thì tỉ lệ cứu sống và phát triển bình thường là 80%.
VI.Tại sao phải sàng lọc sơ sinh?
- Sàng lọc sơ sinh là biện pháp phòng bệnh tích cực cho nhóm bệnh rối loạn chuyển hoá bẩm sinh acid hữu cơ, acid amin, acid béo.
– Nếu được sàng lọc và chẩn đoán bệnh trước khi có triệu chứng xuất hiện thì tỉ lệ cứu sống và phát triển bình thường là 80%
– Nếu được chẩn đoán và điều trị khi có xuất hiện triệu chứng thì tỉ lệ cứu sống và phát triển bình thường có 20%.
- Tư vấn di truyền cho các lần sinh sau và thế hệ sau
– Nhóm bệnh rối loạn chuyển hoá bẩm sinh là bệnh di truyền nên có thể di truyền cho thế hệ sau và các lần sinh sau.
– Tư vấn di truyền giúp chẩn đoán trước sinh, chẩn đoán tiền làm tổ giúp sinh ra những trẻ khoẻ mạnh cho những lần sinh sau.
(Ảnh minh họa. Nguồn: Internet)
VII. Các đối tượng nào nên được làm sàng lọc các bệnh rối loạn chuyển hoá bẩm sinh?
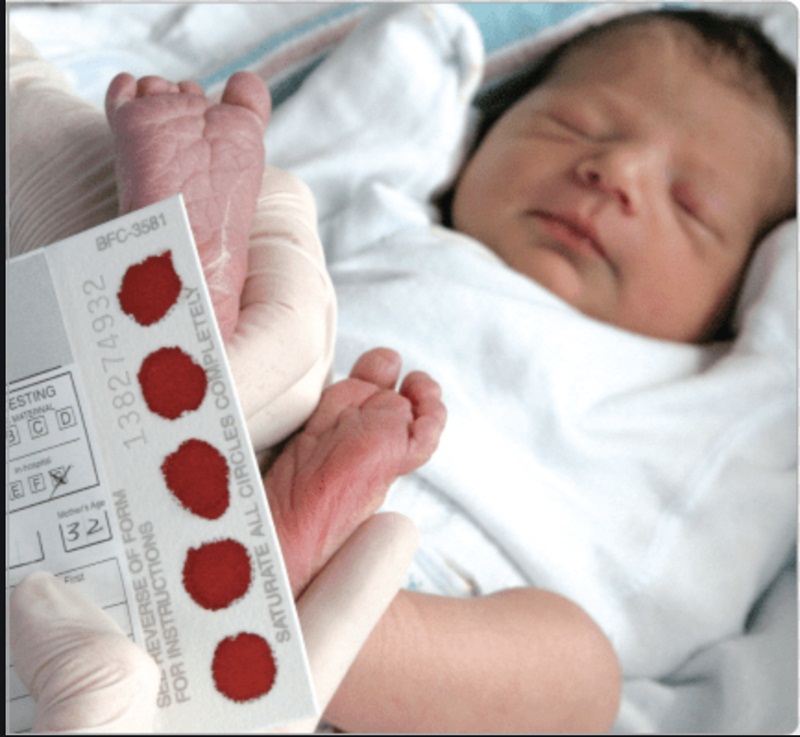
- Tất cả các trẻ sau sinh 24 giờ nên được lấy máu gót chân làm sàng lọc rối loạn chuyển hoá bẩm sinh để phát hiện bệnh sớm khi chưa có biểu hiện lâm sàng.
- Các gia đình có một hoặc nhiều con tử vong không rõ nguyên nhân
- Các trẻ chậm phát triển tâm thần, vận động chưa rõ chẩn đoán
- Các trẻ có biểu hiện như đã liệt kê ở trên.
(Ảnh minh họa. Nguồn: Internet)
DANH SÁCH 55 BỆNH RỐI LOẠN CHUYỂN HOÁ BẨM SINH ACID AMIN, ACID HỮU CƠ, ACID BÉO ĐƯỢC SÀNG LỌC SƠ SINH
Bệnh rối loạn chuyển hóa acid amin (Amino Acid Disorders)
- Argininemia (ARG1 Deficiency)
- Argininosuccinic Aciduria (ASL Deficiency)
- Citrullinemia I (ASS Deficiency)
- Citrullinemia II
- Homocystinuria
- Hypermethioninemia
- Hyperammonemia, Hyperornithinemia, Homocitrullinemia Syndrome1
- Hyperornithinemia with Gyral Atrophy 1
- Maple Syrup Urine Disease
- N-acetyl Glutamate Synthetase Deficiency (NAGS Deficiency)
- Phenylketonuria
- Benign Hyperphenylalaninemia
- Defects of Biopterin Cofactor Biosyntheis
- Defects of Biopterin cofactor regeneration
- Tyrosinemia Type I
- Tyrosinemia Type II
- Tyrosimenia Type III
- Ornitine transcarbamoylase deficiency (OTC)
- Non ketotic hyperglycinemia
- Pyruvate carboxylase deficiency
- Hyperprolinaemia Types I-II (incr. Pro),
- hypoprolinaemia (decr. Pro)
- HHH syndrome (Hyperammonia + Hyperornithinaemia + Homocitrullinuria)
- Hyperlysinaemia, Lysinuric Protein Intolerance (LPI)
- Serine deficiency disorders
Bệnh lý acid hữu cơ (Organic Acid Disorders)
- 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency
- Glutaric Acidemia Type I
- Isobutyryl-CoA Dehydrogenase Deficiency
- Isovaleric Acidemia
- 2-Methylbutryl-CoA Dehydrogenase Deficiency
- 3-Methylcrotonyl-CoA Carboxylase Deficiency
- 3-Methylglutaconyl-CoA Hydratase Deficiency
- Methylmalonic Acidemias
- Some Adenosylcobalamin Synthesis Defects
- Maternal Vitamin B12 Deficiency
- Mitochondrial Acetoacetyl-CoA Thiolase Deficiency (beta-ketothiolase deficiency)
- Propionic Acidemia
- Multiple-CoA Carboxylase Deficiency
- Malonic Aciduria
- Ethylmalonic encephalopathy
Bệnh lý rối loạn chuyển hóa acid béo (Fatty Acid Oxidation Disorders)
- Medium Chain Acyl-CoA Dehydrogenase Deficiency (MCAD)
- Very Long Chain Acyl-CoA Dehydrogenase Deficiency (VLCAD)
- Medium/Short Chain Hydroxy Acyl-CoA Dehydrogenase Deficiency
- 3-Hydroxy Long Chain Acyl-CoA Dehydrogenase Deficiency (LCHAD)
- Short Chain Acyl-CoA Dehydrogenase Deficiency
- Medium Chain Ketoacyl-CoA Thiolase deficiency
- Carnitine uptake deficiency
- Carnitine/Acylcarnitine Translocase Deficiency
- Carnitine Palmitoyl Transferase Deficiency Type II
- Carnitine Palmitoyl Transferase Ia deficiency
- Carnitine Palmitoyl Transferase Ib deficiency
- 2,4-Dienoyl-CoA Reductase Deficiency1
- Glutaric Acidemia type II
- Trifunctional Protein Deficiency
Ethylmalonic encephalopathy
Rối loạn chuyển hoá carbonhydrate
- Galactosemia
Rối loạn dự trữ thể tiêu bào:
- Pompe
- MPS1
- Gaucher
- Fabry
- Niemann Pick
- Krabble
VIII. Vì sao nên đưa trẻ đến Bệnh viện Nhi Trung ương để sàng lọc bệnh lý rối loạn chuyển hoá bẩm sinh?
Bệnh viện Nhi Trung ương là bệnh viện đứng đầu trong cả nước về chẩn đoán và điều trị cho nhóm bệnh rối loạn chuyển hoá bẩm sinh.
- Trung tâm Nội tiết – Chuyển hoá – Di truyền và Liệu pháp phân tử, Bệnh viện Nhi Trung ương đã có 16 năm kinh nghiệm, chẩn đoán và điều trị gần 600 bệnh nhân với 30 loại bệnh rối loạn chuyển hoá bẩm sinh acid amin, acid hữu cơ, acid béo; hơn 200 bệnh nhóm bệnh dự trữ thể tiêu bào gồm 7 týp MPS, Pompe. Trung tâm là nơi đầu tiên trong cả nước điều trị enzyme thay thế cho nhóm bệnh dự trữ thể tiêu bào. Hiện tại Trung tâm Nội tiết – Chuyển hoá – Di truyền và Liệu pháp phân tử, Bệnh viện Nhi Trung ương có số bệnh nhân Pompe, MPS được điều trị enzyme thay thế nhiều nhất khu vực Đông Nam Á.
- Bệnh viện Nhi Trung ương là bệnh viện duy nhất trong cả nước có trang thiết bị hiện đại để làm các xét nghiệm chuyên sâu tiếp theo như phân tích acid hữu cơ niệu và định lượng acid amin máu để xác định chẩn đoán.
- Ngoài ra, bệnh viện còn hợp tác với rất nhiều trung tâm lớn trên thế giới như Nhật Bản, Hoa Kỳ, Đài Loan… để xác định được những khiếm khuyết trên gen gây ra rối loạn hoá bẩm sinh, từ đó tư vấn di truyền cho gia đình bệnh nhân.
- Đội ngũ bác sỹ là các chuyên gia đầu ngành. Năm 2013, 2018, các bác sỹ của khoa đã vinh dự nhận được giải thưởng Eto do Hội rối loạn chuyển hoá bẩm sinh của Nhật Bản trao tặng vì những đóng góp trong nghiên cứu các bệnh lý rối loạn chuyển hoá bẩm sinh. Bên cạnh đó, bệnh viện còn có đội ngũ dinh dưỡng tiết chế chuyên nghiệp tư vấn chế độ ăn, đội ngũ các bác sỹ và kỹ thuật viên phục hồi chức năng chuyên nghiệp cho từng nhóm bệnh rối loạn chuyển hoá bẩm sinh.
Quý khách hàng có thể lựa chọn thăm khám và theo dõi điều trị bệnh lý rối loạn chuyển hoá bẩm sinh tại Trung tâm Nội tiết – Chuyển hoá – Di truyền và Liệu pháp phân tử, Bệnh viện Nhi Trung ương:
– Phòng khám chuyên khoa Nội tiết – Chuyển hoá – Di truyền – Phòng D128 – D130, Khoa Khám bệnh chuyên khoa (Tầng 1, nhà A). Thời gian khám: từ 7h – 16h30 từ thứ 2 đến thứ 6 hàng tuần (trừ ngày lễ, Tết). ĐT: 024 6274 7706.
– Phòng khám chuyên khoa Nội tiết – Chuyển hoá – Di truyền – Trung tâm Quốc tế S. Thời gian khám: từ 7h – 16h30 từ thứ 2 đến thứ 6 hàng tuần và từ 7h – 12h sáng thứ 7 hàng tuần (trừ ngày lễ, Tết). ĐT: 024 6273 8900.
Khoa Nội tiết – Chuyển hóa – Di truyền
Bệnh viện Nhi Trung ương
![[Thông báo mời báo giá] Về việc thẩm định giá, xác định giá tài sản công cụ dụng cụ chờ thanh lý tại Bệnh viện Nhi Trung ương](https://benhviennhitrunguong.gov.vn/wp-content/uploads/2026/07/TB-Mua-sam_366x236-150x150.png)



